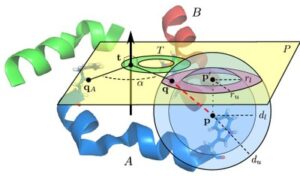
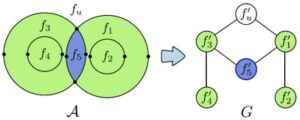
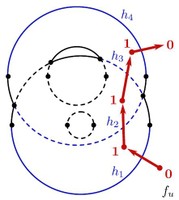
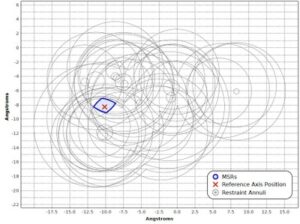
Nuclear magnetic resonance (NMR) spectroscopy is a primary tool to perform structural studies of proteins in physiologically-relevant solution conditions. Restraints on distances between pairs of nuclei in the protein, derived from the nuclear Overhauser effect (NOE), provide information about the structure of the protein in its folded state. NMR studies of symmetric protein homo-oligomers present a unique challenge. Using X-filtered NOESY experiments, it is possible to determine whether an NOE restrains a pair of protons across different subunits or within a single subunit, but current experimental techniques are unable to determine in which subunits the restrained protons lie. Consequently, it is difficult to assign NOEs to particular pairs of subunits with certainty, thus hindering the structural analysis of the oligomeric state. Computational approaches are needed to address this subunit ambiguity, but traditional solutions often rely on stochastic search coupled with simulated annealing and simulations of simplified molecular dynamics, which have many tunable parameters that must be chosen carefully and can also fail to report structures consistent with the experimental restraints. In addition, these traditional approaches rarely provide guarantees on running time or solution quality. We reduce the structure determination of homo-oligomers with cyclic symmetry to computing geometric arrangements of unions of annuli in a plane. Our algorithm, DISCO, runs in expected \(O(n^2)\) time, where \(n\) is the number of distance restraints, potentially assigned ambiguously. DISCO is guaranteed to report the exact set of oligomer structures consistent with the distance restraints and also with orientational restraints from residual dipolar couplings (RDCs). We demonstrate our method using two symmetric protein complexes: the trimeric E. coli Diacylglycerol Kinase (DAGK), and a dimeric mutant of the immunoglobulin-binding domain B1 of streptococcal protein G (GB1). In both cases, DISCO computes oligomer structures with high precision and also finds distance restraints that are either mutually inconsistent or inconsistent with the RDCs